How to guide: Clustering tool
Tool view

Purpose of tool
The tool performs clustering on localisation data to find biological structures and quantify their statistical and morphological properties. The default clustering method is HDBSCAN. The cluster results can then be dynamically refined with DBSCAN* or constrained based on cluster features related to the morphology of the clusters.
Contents
Running the tool
Inspecting outputs
Viewing results
Inspecting analysis results
Optional: Refinement of clustering results
Constrained clustering
DBSCAN*
Exporting results
Saving raw cluster data
Saving histograms
Running the tool
Clustering can be run with default parameters by clicking “run” in the widget. This will apply any pre-processing steps loaded in the workflow to the localisations before clustering each channel individually. For more fine-grained control, a menu can be accessed by clicking the three horizontal line icon to reveal further options.
For multi-channel datasets, it is possible to select which channels to cluster as well as whether to cluster them together (when “Merged” is highlighted) or separately.
The following HDBSCAN parameters can also be selected before clustering:
Minimum samples (Min samples)
This is related to the minimum density of the clusters
Minimum number of localisations per cluster (Min size)
This sets a lower limit on the size of a cluster
Previous clustering executions can also be selected using the load button. Once a clustering execution has finished it should automatically load in the clustering results from the first ROI and channel (if multiple are present, the user can toggle between them).
Current and previous clustering executions can also be viewed and selected by clicking on the load button.
Inspecting outputs
Once the results are loaded, a new layer is added for the clusters as shown below. Each cluster is shown in a different colour by default, the “Shuffle colors” button randomly reorders these colors.
There is also the option to show or hide noise points in the visualisation. These are points that the algorithm has not placed inside a cluster.
Only one channel-mask combination can be viewed at once.
Cluster features
From this widget the user can inspect the distribution of each cluster feature, toggle between the different ROIs and channels, refine the clusters and export the data. The widget also displays the total number of clusters found.
The following cluster features are calculated:
Number of localisations
Skew - The ratio of the major to the minor axis
Circularity - A measure where 1 corresponds to a circle, and lower values are assigned to more irregularly shaped clusters
Density - Number of localisations in a cluster divided by the area of the cluster
Area - Computed from the convex hull of the cluster
Discretised area - A measure of a cluster’s area based on binning its localisations into pixels
Length - An approximation of the longest axis
Kappa - model parameter relating to the least dense parts of a cluster.
More details are available in the technical document on clustering.
Refinement of clustering results
Clustering methods rely on statistical measures to detect features in an image. However, the features of interest for a particular biological analysis may not be the most statistically significant and thus not found by the initial clustering. For example, it is possible that the structures of interest in the image are circular but the clustering algorithm will not, by default, look for circular clusters. In such situations, constraining the clusters based on geometric/morphological features can be useful. The clustering tool enables this by providing two further tools for further refinement of clusters.
Constrained clustering
This approach allows the user to set constraints on the cluster properties and then recluster the data according to these constraints.
DBSCAN*
This approach is similar to DBSCAN but allows for real time adjustment of the eps parameter to choose a cluster length scale.
NOTE: The graphs disappear when the refinement process is started. The original clustering results are still available from the “Load” menu, and the “revert” button will undo any changes. Once the refinement is complete, click “save to cloud” to compute the new feature distribution graphs and/or to use the clusters for a subsequent step in the workflow.
Constrained clustering
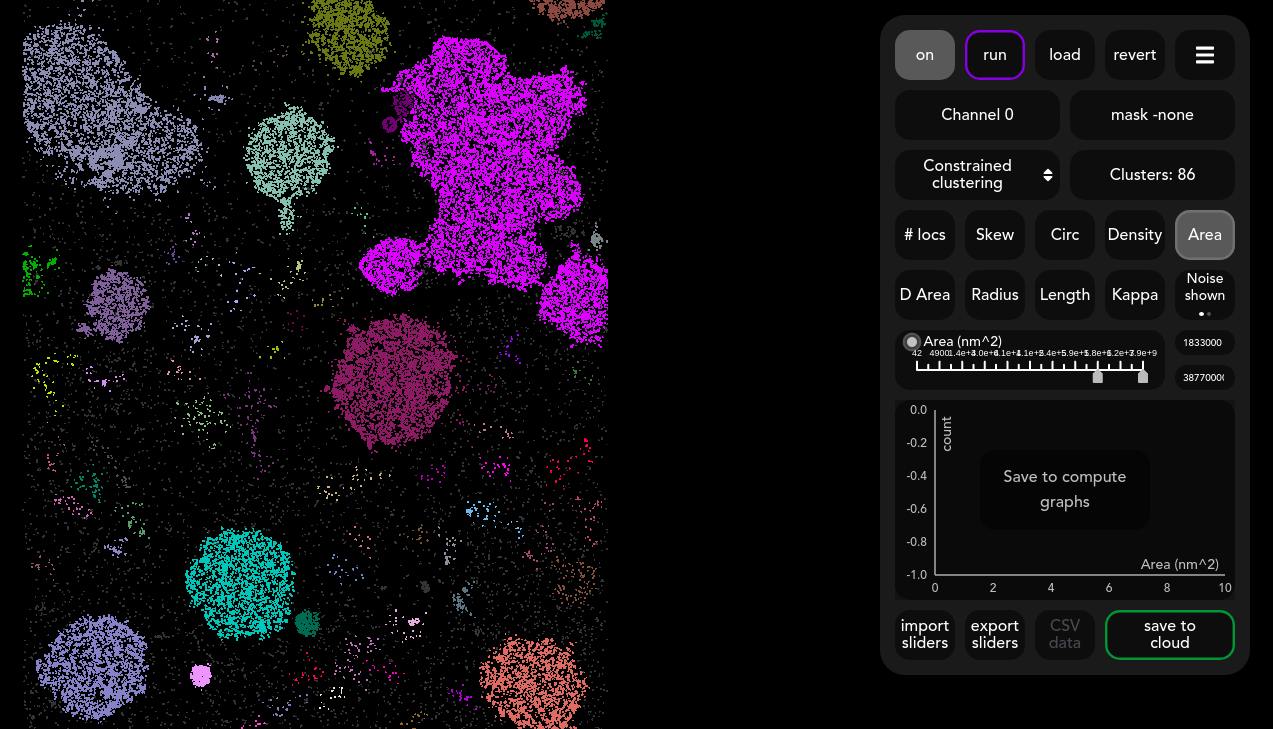
For each feature, the sliders can be adjusted to restrict the type of clusters being computed. The algorithm will then dynamically recluster the localisations to fit these constraints. In this case, a minimum area threshold has been selected, which has caused smaller clusters to coalesce into larger ones. Multiple constraints can be selected, and a grey outline is shown around those that are active. A quick way to see the effect of an adjusted constraint is to click the toggle on the slider on/off for that feature.
The slider ticks have been selected such that it should be easy to choose the right constraints but it is also possible to input specific numbers by hand in the text boxes on the right of the slider.
DBSCAN*
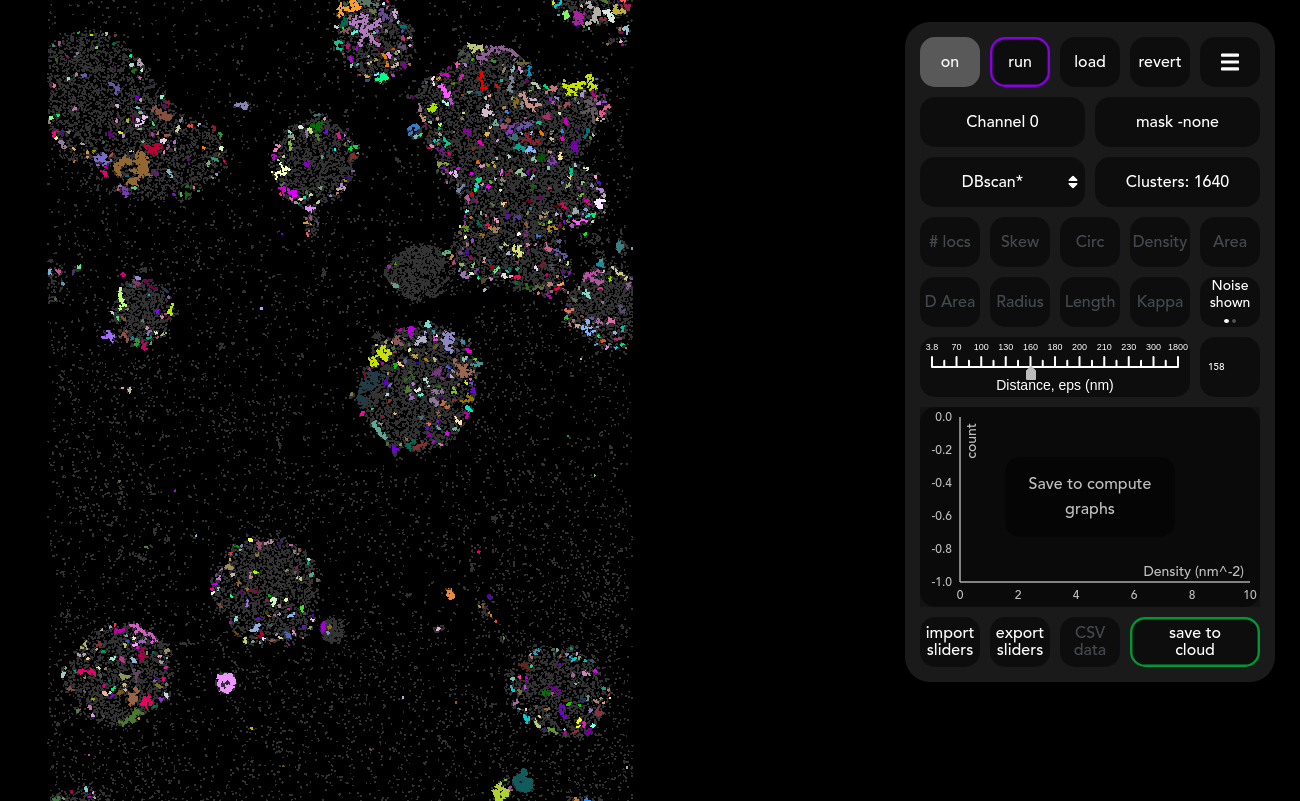
An alternative to constrained clustering is to use DBSCAN*. This refinement method only has one slider ‘Distance’ which mimics the behaviour of the DBSCAN eps parameter (more info). Move this slider to change the length scale of the clusters. The effect of the increasing the slider can be seen in the image below.
Larger values of eps allows for a larger separation of points within each cluster, which typically leads to less dense and larger clusters.
Exporting results
Saving raw cluster data
The “CSV data” button provides a CSV where each row corresponds to an individual cluster. The CSV contains the ID of each cluster, as well as each cluster feature.
Saving histograms
Each of the histograms can be downloaded as an image or the raw data can also be downloaded as a CSV. Use the download button in the top right corner of the graph.
An example of the CSV file for the skew histogram is given below:
Related Articles
How to guide: Counting tool
Tool view Purpose of tool Counts the number of localisations or ‘bins’ by channel around a set of centre points - currently the centroids of a clustering result. Contents Inputs Results Outputs Inputs The centre points for counting come from a loaded ...How to guide: Filtering tool
Tool view Purpose of tool Enable user to filter out points based on their properties and save the output for further analysis in workflow. Contents Input parameters Multiple channels Output results Input parameters The points in dSTORM acquisitions ...How-to-guide: Temporal grouping tool
Tool view Purpose of tool In localisations-based super-resolution microscopy, each fluorescent molecule can emit ("blink") multiple times, depending on its properties and the super-resolution technique used. Those blinking events are localized ...August 2023 Clustering User Interface Improvement
While optimizing CODI during its beta phase, we noticed some inconsistencies in the clustering widget when using the DBSCAN* algorithm, whereby the CODI user interface misrepresents information pertaining to number of clusters and localizations. Data ...How to guide: Annotation tool (ROI)
Purpose of tool Select regions of interest (ROIs) in images. How to draw ROIs To begin drawing an ROI, click on one of the three green button depending on the shape you would like to draw. For the polygon: Click on the image, if possible around the ...